Malá změna v kitu SALSA® MLPA® probemix P064-C2 Microdeletion Syndromes-1B
Katalogová čísla kitu jsou:
- P064-025R: SALSA® MLPA® probemix P064 Microdeletion Syndromes-1B, 25 reakcí.
- P064-050R: SALSA® MLPA® probemix P064 Microdeletion Syndromes-1B, 50 reakcí.
- P064-100R: SALSA® MLPA® probemix P064 Microdeletion Syndromes-1B, 100 reakcí.
Kit používejte v kombinaci s reagenčním SALSA® MLPA® kitem dostupným pro různý počet reakcí. MLPA reagenční kity máte k dispozici s FAM nebo Cy5.0 značenými primery. Nový kit je také vhodný pro kapilární sekvenátory Applied Biosystems a Beckman.
Kit je určený pouze pro profesionální použití. Vždy si přečtěte nejnovější popis produktu a MLPA obecný protokol. Je vaší odpovědností, abyste se seznámili s nejnovějšími vědeckými poznatky, než budete vyvozovat závěry z výsledků. Informace o podmínkách skladování, testech kvality a vzoru elektroforeogramu z aktuální šarže máte k dispozici na adrese mlpa.com.
Využití nové verze kitu
Kit SALSA® MLPA® probemix P064 Microdeletion Syndromes-1B je učen pro in vitro diagnostické (IVD) testy nebo jen pro výzkumné účely (RUO).
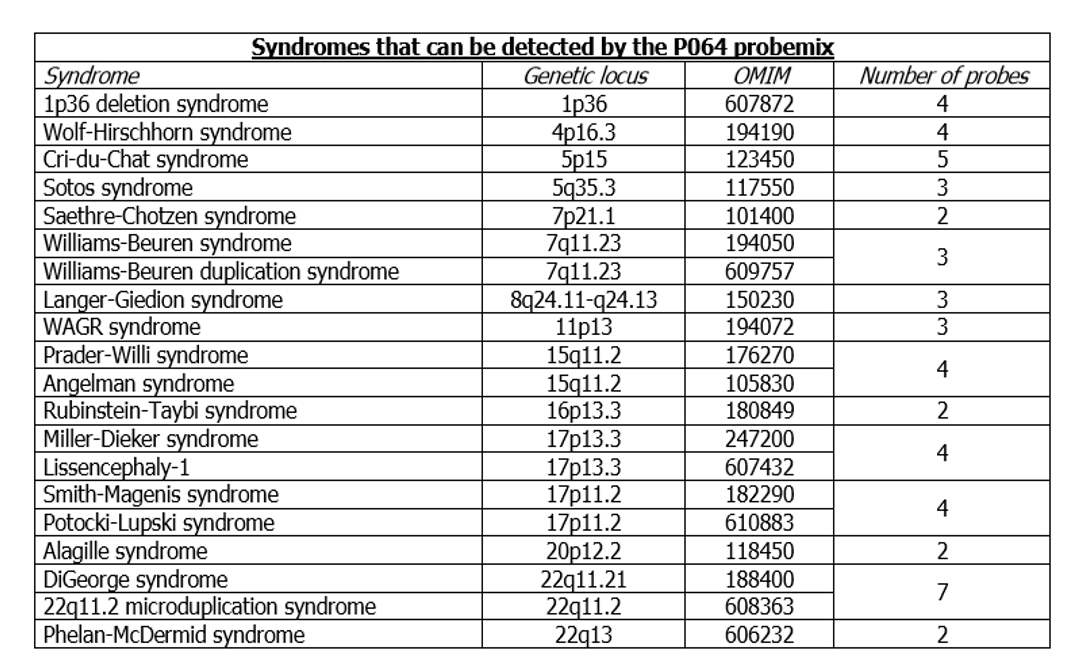 pro detekci odlišné podskupiny rekurentních mikrodelecí a mikroduplikací v lidské DNA izolované z periferní krve,
pro detekci odlišné podskupiny rekurentních mikrodelecí a mikroduplikací v lidské DNA izolované z periferní krve,- bukálního stěru,
- (ne)kultivované plodové vody získané v 16. týdnu těhotenství nebo později a bez kontaminace krve,
- (ne)kultivovaných choriových klků bez maternální kontaminace nebo krve plodu, aby se potvrdila příčina a klinická diagnóza pro vývojové zpoždění nebo syndromy intelektuálního postižení.
Klinické pozadí
Mikrodeleční a mikroduplikační syndromy je skupina klinicky rozlišitelných onemocnění charakterizovaných malou (<5Mb) delecí nebo duplikací chromozomálního segmentu zahrnujícího více patologických genů. Fenotyp je výsledkem haploinsuficience specifických genů v kritickém intervalu.
Klinicky dobře popsané syndromy, u kterých bylo zjištěno zapojení mnoha genů:
- DiGeorgeův syndrom (mikrodelece 22q11),
- WilliamsBeuren syndrom (mikrodelece 7q11),
- Smith-Magenisův syndrom (17p mikrodelece) a mnoho dalších.
Postihují 1-3 % populace a vznikají z mimořádných heterogenních enviromentálních, chromozomálních a monogenních příčin.
Jednu z nejobtížnějších výzev, kterým dnes čelí lékaři a genetici, představuje intelektuální postižení (ID). Podrobná analýza databáze Online Mendelian Inheritance in Man a literární výzkum odhalily více než tisíc záznamů o ID a více než 290 genů zodpovědných za klinický fenotyp nebo syndromy, metabolické nebo neurologické poruchy charakterizovaných ID. Genetické změny mikrodelecí/duplikací často nejsou detekovatelné běžným rozlišením pruhů používající rutinní nebo vysoce rozlišovací karyotypování (2-5 Mb), ale vyžadují použití molekulárně cytogenetických technik, jako je fluorescence in situ hybridizace (FISH), MLPA nebo array komparativní genomová hybridizace (aCGH).
Obsah nové verze P064-C2 kitu
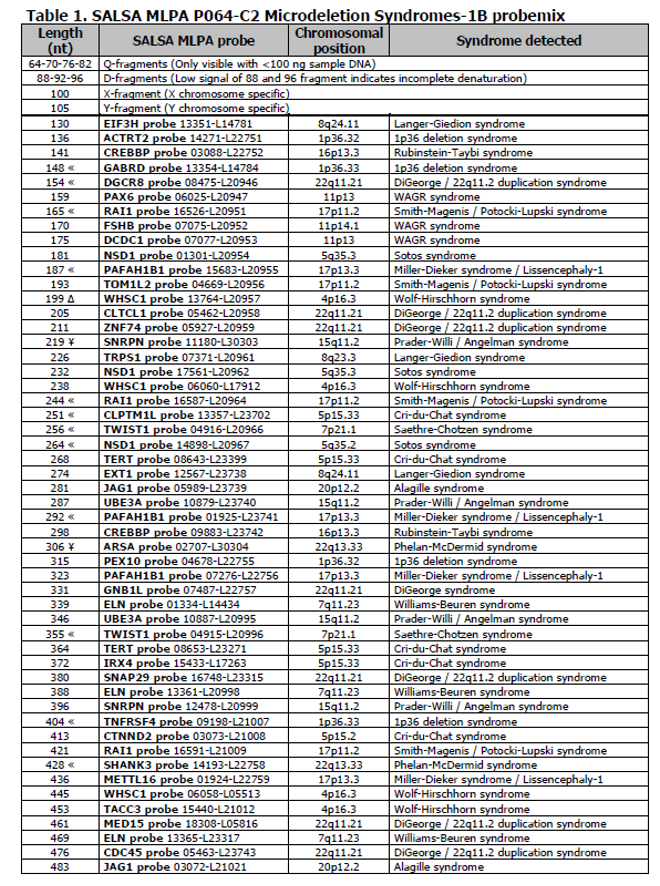
Kit SPA MLPA P064 Microdeletion Syndromes-1B obsahuje 52 MLPA sond s produkty amplifikace mezi 130 a 483 nt. Sondy detekují sekvence podílející se na odlišné podskupině mikrodelečních a mikroduplikačních chorob.
Tento kit obsahuje devět kontrolních fragmentů, které generují amplifikační produkty mezi 64 a 105 nt:
- čtyři DNA kvantitativní fragmenty (Q-fragmenty),
- tři DNA denaturační fragmenty (D-fragmenty),
- jeden chromozóm X,
- jeden chromozóm Y-specifický fragment (Tabulka 1).
Q-fragmenty jsou viditelné pouze, pokud je použito méně než 100 ng DNA vzorku. Nízký signál 88 nebo 96 nt fragmentu naznačuje neúplnou DNA denaturaci.
Validace MLPA metodyInterní validace MLPA metody na 16 vzorcích DNA ze zdravých jedinců je nezbytná, a to zejména při prvním použití MLPA, nebo při změně zpracování vzorků, metodě extrakce DNA nebo používaného přístroje. Validace by měla vykazovat standardní odchylku <0,10 pro všechny sondy v experimentu. |
Požadavky na vzorkyDNA izolovaná z periferní krve, bukálního stěru, (ne)kultivované plodové vody získané v 16. týdnu těhotenství nebo později a bez kontaminace krve, (ne)kultivovaných choriových klků bez maternální kontaminace nebo krve plodu, bez nečistot, které ovlivňují MLPA reakci. |
Referenční vzorkyReferenční vzorky DNA odvozujte ze stejného typu tkáně, zpracujte je za použití stejného postupu a připravte za použití stejného způsobu, jako je například extrakce DNA ze vzorku pacienta. Referenční vzorky by měly být odvozeny od nepříbuzných jedinců, kteří jsou z rodin bez historie syndromů opožděného vývoje a/nebo intelektuálního postižení. |
Pozitivní kontrolní vzorky DNAPamatujte, že MRC-Holland nemůže poskytnout pozitivní vzorky DNA. Zahrnutí pozitivních vzorků se doporučuje v každém experimentu. Pozitivní kontroly mohou být objednány z Coriell Institutu pro většinu syndromů zahrnutých v tomto kitu. Berte, prosím, na vědomí, že tyto vzorky nebyly testovány v MRC Holland. |
Charakteristika výkonu testu
Klinický význam nové verze kitu závisí hlavně na sledovaných populacích nebo kohortách. Podle literatury přibližně 10 až 20 % vzorků pacientů testovaných pomocí kitu P064 vykazuje mikrodelece nebo mikroduplikace, což vede k významnému diagnostickému výtěžku při testování syndromů intelektuálního postižení a/nebo chromozomální nerovnováhy.
Analytickou výkonnost můžete ohrozit:
-
SNP nebo dalšími polymorfismy (např. indels) v cílové sekvenci DNA,
MLPA obecný protokol obsahuje technické pokyny a informace o hodnocení a normalizaci dat.
- nečistotami ve vzorku DNA,
- neúplnou DNA denaturací,
- použitím nedostatečného nebo příliš velkého množství vzorku DNA,
- použitím nedostatečných nebo nevhodných referenčních vzorků,
- problémy s kapilární elektroforézou nebo špatným postupem normalizace dat a dalšími technickými chybami.
Coffalyser.Net software použijte pro analýzu dat v kombinaci s listem pro specifickou šarži - MLPA Coffalyser list. Použití jiného ne-proprietárního softwaru může vést k neprůkazným nebo falešným výsledkům.
Interpretace výsledků
Očekávané výsledky pro sondy jsou počty kopií alel 2 (normální), 1 (heterozygotní delece), nebo 3 (heterozygotní duplikace). Ve vzácných případech může být počet kopií 0 (homozygotní delece) nebo 4 (heterozygotní triplikace/homozygotní duplikace).
Standardní odchylka všech sond v referenčních vzorcích by měla být <0,10 a kvocient dávky (DQ) referenčních sond ve vzorcích pacientů by měl být mezi 0,80 a 1,20. Pokud jsou tato kritéria splněna, následující mezní hodnoty pro DQ sond můžete použít k interpretaci výsledků MLPA.
 Uspořádání sond podle chromozomální polohy vám usnadní interpretaci výsledků a může odhalit menší změny, jako jsou změny pozorované v případech mozaiky. Analýza rodičovských vzorků může být nezbytná pro správnou interpretaci komplexních výsledků.
Uspořádání sond podle chromozomální polohy vám usnadní interpretaci výsledků a může odhalit menší změny, jako jsou změny pozorované v případech mozaiky. Analýza rodičovských vzorků může být nezbytná pro správnou interpretaci komplexních výsledků.
Vezměte prosím na vědomí, že abnormality zjištěné jedinou sondou (nebo více po sobě jdoucích sond) mají stále značnou šanci mít falešně pozitivní výsledek. Neúplná DNA denaturace (například z důvodu kontaminace solí) může vést ke snížení signálu sondy, zejména u sond umístěných v CpG ostrově. Použití dalšího kroku čištění nebo alternativní metody extrakce DNA může takové případy vyřešit.
Falešné pozitivní výsledky - duplikace
Kontaminace DNA vzorků cDNA nebo PCR amplikony z jednotlivých exonů mohou vést k falešně pozitivním duplikacím (Varga et al., 2012). Analýza nezávisle odebraného vzorku sekundární DNA může vyloučit tento typ artefaktů. Normální počty kopií variant u zdravých jedinců jsou popsány v databázi genomových variant. Vždy konzultujte nejnovější aktualizaci databáze a vědecké poznatky při interpretaci jejich výsledků.
Ne všechny odchylky zjištěné MLPA jsou patogenní. V některých genech, jsou známy intragenové delece a vedou k velmi mírné nebo žádné chorobě (Schwartz et al., 2007). Pro mnoho genů existuje více než jedna transkripční varianta. Změny v počtu kopií exonů, které nejsou přítomny ve všech transkripčních variantách, nemusí mít klinický význam. Duplikace, které zahrnují první nebo poslední exon genu (například exony 1-3), mohou v některých případech mít za následek inaktivaci této kopie genu. Změny v počtu kopií detekované referenčními sondami nebudou mít pravděpodobně žádný vztah k testovanému onemocnění.
Limitace metody
- P064 kit má omezené množství sond pro každou specifickou chromozomální oblast, a proto nezjistí všechny možné příčiny zahrnutých syndromů. Míra detekce se může mezi syndromy lišit v závislosti na heterogenitě choroby.
- U syndromů Prader-Willi a Angelman lze kit P064 použít pouze k detekci změn počtu kopií v oblasti 15q11.2. Sondy pro detekci změn metylace v tomto místě jsou přítomny v kitu ME028.
- MLPA nemůže zjistit žádné změny, které leží mimo cílovou sekvenci sond a nezjistí počet kopií neutrální inverze nebo translokace. I když MLPA nedetekuje žádné aberace, zůstává možnost, že existují biologické změny v tomto genu nebo chromozomální oblasti, ale zůstávají nedetekované.
- Změny sekvence (např. SNP, bodové mutace, malé indels) v cílové sekvenci sondy mohou způsobit falešně pozitivní výsledky. Mutace / SNP (i když> 20 nt od ligačního místa sondy) může snižovat signál sondy tím, že brání ligaci oligonukleotidové sondy nebo destabilizuje vazbu sondy na DNA.
Potvrzení výsledků
Změny v počtu kopií detekované pouze jedinou sondou vždy vyžadují ověření jinou metodou. Zřejmá delece detekovaná jedinou sondou může být způsobena např. mutací/polymorfismem, který zabraňuje ligaci nebo destabilizuje vazbu sondy na DNA. Sekvenační analýza může stanovit, zda mutace nebo polymorfismy jsou přítomny v cílové sekvenci sondy. Nalezení heterozygotní mutace nebo polymorfismu poukazuje na to, že dvě odlišné alely jsou přítomné ve vzorku DNA a lze získat falešně pozitivní výsledek MPLA.
Změny v počtu kopií detekované jednou nebo více než jednou po sobě jdoucích sond by měly být potvrzeny jinou nezávislou metodou, jako je long-range PCR, qPCR, array CGH nebo Southern blotting, kdykoli je to možné. Delece/duplikace delší než 50 kb mohou být často potvrzeny FISH. Dále mohou být potvrzeny změny v počtu kopií zjištěné kitem P064 pomocí syndrom - specifického kitu MLPA. Téměř pro všechny syndromy velký počet sond pro dané chromozomální oblast (i) je zahrnut v kitech P371, 372, P373 a P374.
Databáze DECIPHER
Důrazně doporučujeme uživatelům ukládat pozitivní výsledky do databáze DECIPHER. Doporučení pro názvosloví, jak popisovat delece/duplikace jednoho nebo více exonů lze nalézt zde. Prosím, oznamte nám falešně pozitivní výsledky kvůli SNP a neobvyklé výsledky (například duplikace dvou sond, které nejsou za sebou v pořadí).
 «Sonda umístěná uvnitř nebo blízko ostrova CpG. Nízký signál může být způsoben kontaminací solí ve vzorku DNA, což vedlo k neúplné denaturaci DNA, zvláště v oblastem bohatých na CG.
«Sonda umístěná uvnitř nebo blízko ostrova CpG. Nízký signál může být způsoben kontaminací solí ve vzorku DNA, což vedlo k neúplné denaturaci DNA, zvláště v oblastem bohatých na CG.
Δ Více vaiabilní. Tato sonda může být citlivá na určité experimentální změny. Aberantním výsledkům by měla být věnováná zvýšená pozornost.
¥ Změněno ve verzi C2 (od šarže C2-0817 a dále). Malá změna v sekvenci, žádná změna v místě ligace.
Sondy pro potvrzení jsou k dispozici v kitu P373 Microdeletion Syndromes 7. SALSA MLPA kit P147 obsahuje více sond zaměřených na sekvenci 1p36. Delece v oblasti 1p36 byly hlášeny jako častá příčina vývojového zpoždění a intelektuálního postižení s frekvencí od 1:5 000 do 1:10 000 porodů. Většina případů zahrnuje terminální delece, které by měly být také detekovány kity P036 a P069 / P070 SALSA MLPA. Bylo popsáno několik intersticiálních delecí a komplexních přestaveb.
Více informací o delečním syndromu 1p36 lze nalézt tady. Pacienti s delečním syndromem 1p36 mají typické kraniofaciální znaky, brachy (camptodactylii), krátké nohy a vývojové zpoždění a intelektuální postižení různého stupně. Hypotonie, záchvaty, strukturální abnormality mozku a vrozené srdeční vady.
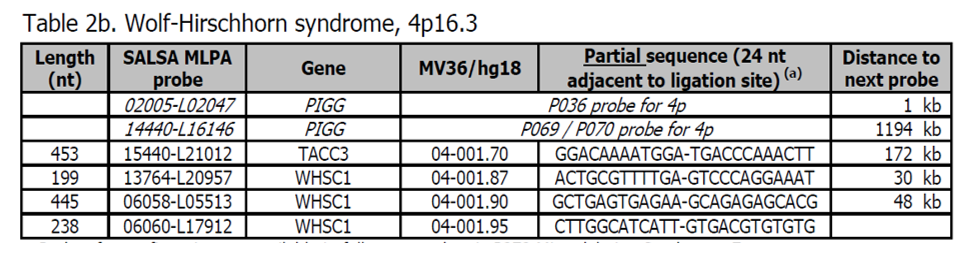 Sondy pro potvrzení jsou k dispozici v kitu P373 Microdeletion Syndromes 7. Další sondy pro oblast Wolf-Hirschhorn jsou přítomny v kitu P096 MR2. Nejčastější příčinou je terminální delece 4p16.3, která může být také detekována telomerickými kity P036 a P069/P070.
Sondy pro potvrzení jsou k dispozici v kitu P373 Microdeletion Syndromes 7. Další sondy pro oblast Wolf-Hirschhorn jsou přítomny v kitu P096 MR2. Nejčastější příčinou je terminální delece 4p16.3, která může být také detekována telomerickými kity P036 a P069/P070.
Kritická oblast WHS se nachází přibližně 1,9 Mb od telomery a zahrnuje gen WHSC1. Více informací o Wolf-Hirschhorn syndromu lze nalézt zde. Fenotyp zahrnuje variabilní míru vývojového zpoždění a intelektuálního postižení, záchvaty a anomálie skeletu.
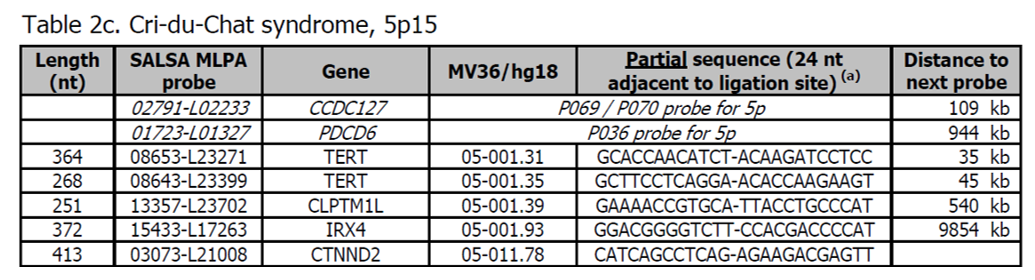
- Sondy pro potvrzení jsou k dispozici v kitu P373 Microdeletion Syndromes 7.
- Další sondy pro oblast Cri-du-Chat jsou přítomny v kitu P096 MR2.
- Nejčastější příčinou Cri-du-Chat syndromu je terminální delece 5p15, která může být také detekována telomerickými kity P036 a P069 / P070. Také byly popsány intersticiální delece (Zhang X et al., (2005) Am JHum Genet 76: 312 - 326).
- Další informace o Cri-du-Chat syndromu naleznete v OMIM 123450. Klinické znaky zahrnují závažnou psychomotorickou retardaci, intelektuální postižení a charakteristický kočičí pláč.
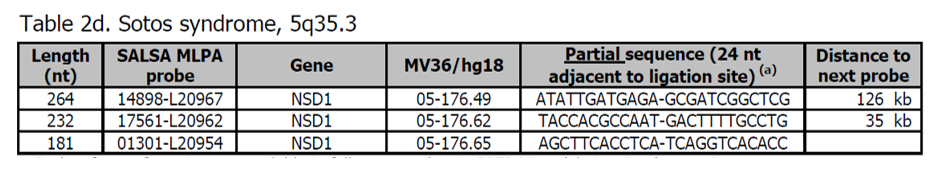 Sondy pro potvrzení máte k dispozici v kitu P372 Microdeletion Syndromes 6. Více sond pro gen NSD1 je přítomno v kitu P026 Sotos syndrom. Ze všech nalezených NSD1 mutací ~ 10% (ne-japonská populace) až ~ 45% (japonská populace) představují kompletní deleci genu. Reciproké duplikace způsobují opačný fenotyp syndromu Sotos (Franco LM et al. (2010) Eur J Hum Genet 18: 258 - 261).
Sondy pro potvrzení máte k dispozici v kitu P372 Microdeletion Syndromes 6. Více sond pro gen NSD1 je přítomno v kitu P026 Sotos syndrom. Ze všech nalezených NSD1 mutací ~ 10% (ne-japonská populace) až ~ 45% (japonská populace) představují kompletní deleci genu. Reciproké duplikace způsobují opačný fenotyp syndromu Sotos (Franco LM et al. (2010) Eur J Hum Genet 18: 258 - 261).
Vzdálenost od genu NSD1 k telomerickým sondám 5q v P036 a P069 / P070 je přibližně 3950 kb. Sotos syndrom je způsoben hlavně bodovými mutacemi v genu NSD1, které nebudou detekovány MLPA. Gen NSD1 je také zapojen do Beckwith-Wiedemannova syndromu, který nemůže být detekován pomocí kitu P064.
Více informací o syndromu Sotos naleznete zde. Sotos syndrom je charakterizován nadměrným fyzickým růstem v kojeneckém věku a makrocefalem a může být doprovázen autismem, mírným intelektuálním postižením a zpožděným motorickým vývojem.
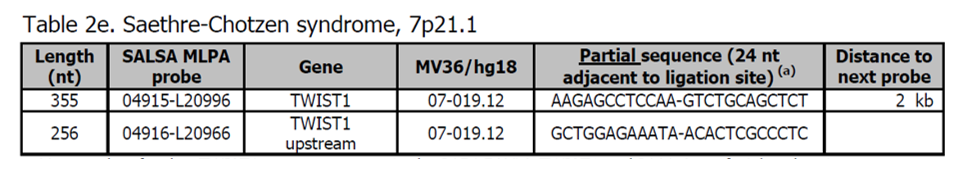
- Další sondy pro gen TWIST1 jsou přítomny v kitech P054 FOXL2-TWIST1 a P080 Cranofacial.
- Většina Saethre-Chotzen syndromů je způsobena bodovými mutacemi v genu TWIST1, které nemohou být detekovány MLPA. Přibližně 11-28 % pacientů s Saethre-Chotzen má deleci zahrnující gen TWIST1.
Více informací o syndromu Saethre-Chotzen naleznete zde a v OMIM 101400. Fenotyp zahrnuje koronální synostózu, obličejovou dysmorfologii a syndactylii. Na rozdíl od pacientů s bodovou mutací TWIST1, pacienti s (částečnou) delecí TWIST1 jsou často vývojově zpožděni.
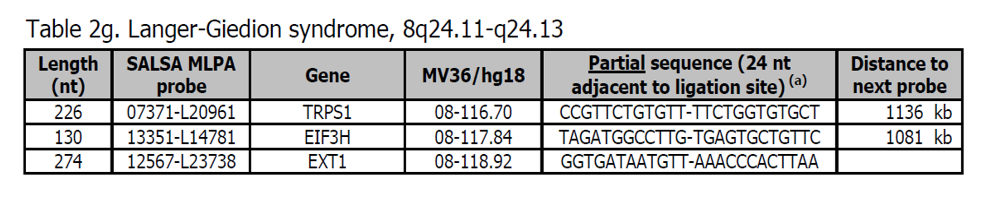 Sondy pro potvrzení jsou k dispozici v kitu P371 Microdeletion Syndromes 5. Další sondy pro oblast Langer-Giedion jsou přítomny v kitech P215 EXT a P228 TRPS1-EXT1. Většina pacientů s Langer-Giedionovým syndromem (LGS) má mikrodeleci, která zahrnuje geny TRPS1 a EXT1. LGS je také známý jako trichorhinofalangeální syndrom typu II (TRPS2).
Sondy pro potvrzení jsou k dispozici v kitu P371 Microdeletion Syndromes 5. Další sondy pro oblast Langer-Giedion jsou přítomny v kitech P215 EXT a P228 TRPS1-EXT1. Většina pacientů s Langer-Giedionovým syndromem (LGS) má mikrodeleci, která zahrnuje geny TRPS1 a EXT1. LGS je také známý jako trichorhinofalangeální syndrom typu II (TRPS2).
Více informací o LGS najdete v OMIM 150230. Fenotyp zahrnuje mnoho dysmorfických obličejových vlastností, vícenásobné chrupavkové exostózy, redundantní kůži, řídké vlasy na pokožce hlavy a mírné až středně těžké mentální postižení.
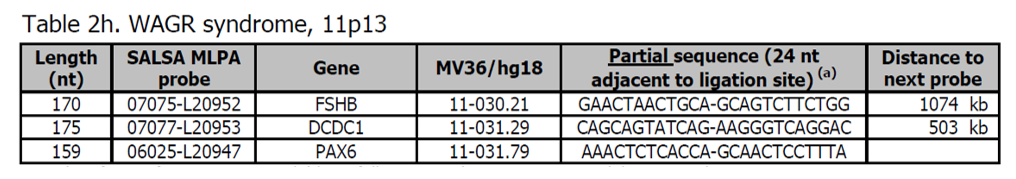
- Sondy pro potvrzení jsou k dispozici v kitu P371 Microdeletion Syndromes 5.
- Více sond pro PAX6 je přítomno v kitu P219 PAX6.
Více informací o WAGR syndromu naleznete v OMIM 194072. Děti postižené syndromem WAGR mají predispozici k rozvoji Wilmsových nádorů, Aniridia, geniturinárních anomálií (nebo gonadoblastomu) a mentálního opoždění.
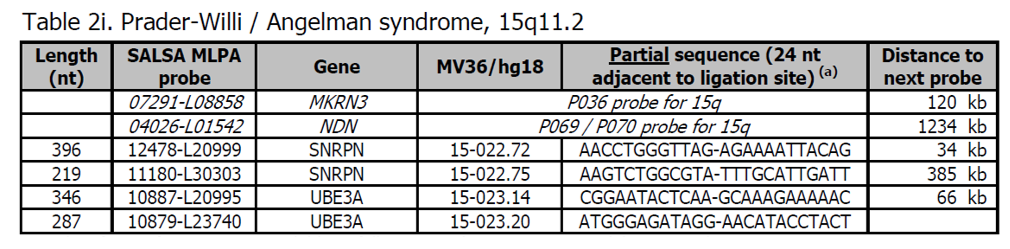
- Sondy pro potvrzení jsou k dispozici v kitu P374 Microdeletion Syndromes 8.
- Více sond pro oblast Prader-Willi / Angelman, včetně sond pro detekci změn metylace, je v kitu ME028.
- Většina pacientů s Prader-Willi a Angelmanem má změnu počtu kopií oblasti 15q11.2, která by měla být detekována tímto P064 kitem. Značný počet pacientů (~ 30%) však vykazuje změnu metylačního stavu oblasti 15q11.2, která může být detekována kitem ME028, ale nikoliv s tímto kitem P064.
Prader-Willi syndrom (PWS) a Angelman syndrom (AS) jsou klinicky odlišné komplexní choroby. Oba syndromy mají charakteristické neurologické, vývojové a behaviorální fenotypy plus další strukturální a funkční abnormality. Kognitivní a neurologické poškození je u AS závažnější, včetně záchvatů a ataxie. Chování a endokrinní poruchy jsou více u PWS, včetně obsedantně-kompulzivních příznaků a hypotalamické nedostatečnosti.
Další informace o syndromu Prader-Williho (PWS) naleznete zde a v OMIM 176270. Více informací o Angelmanově syndromu (AS) najdete tady a v OMIM 105830.
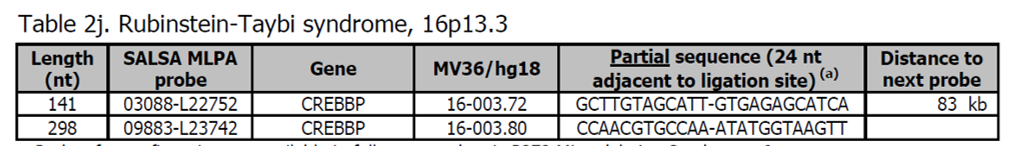
- Sondy pro potvrzení jsou k dispozici v kitu P372 Microdeletion Syndromes 6.
- Další sondy pro gen CREBBP jsou přítomny v kitu P313 CREBBP.
- 16p13.3 deleční syndrom (OMIM 610543) je způsoben většími delecemi, které zahrnují gen CREBBP a způsobují těžkou formu syndromu Rubinstein-Taybi.
Použitím těchto dvou sond může být detekována pouze menšina pacientů s Rubinstein-Taybi (~ 10%), protože většina pacientů má bodovou mutaci v genu CREBBP nebo EP300.
Více informací o syndromu Rubinstein-Taybi lze nalézt zde a v OMIM 180849. Fenotyp zahrnuje výrazné rysy obličeje, široké a často vykulené palce a velké prsty, krátké postavení a středně těžké až těžké intelektuální postižení.
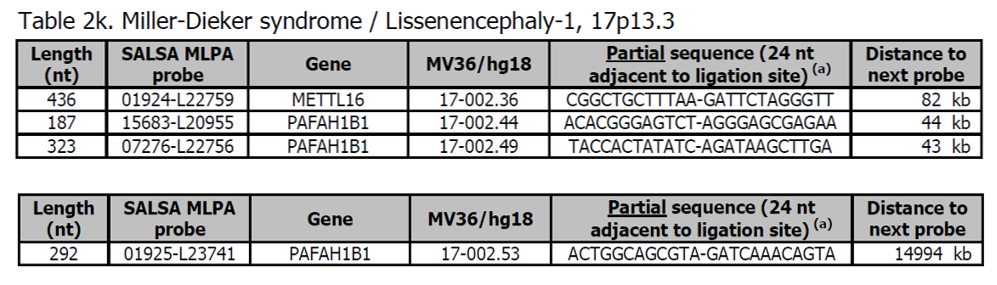
- Sondy pro potvrzení jsou k dispozici v kitu P374 Microdeletion Syndromes 8.
- Více sond pro PAFAH1B1 a další geny v oblasti Miller-Dieker je přítomno v kitu P061 Lissencephaly.
- Většina pacientů s Lissencephaly-1 a téměř všichni pacienti s Miller-Dieker mají chromozomovou deleci, která zahrnuje gen PAFAH1B1. Bylo popsáno několik pacientů s duplikací v oblasti Miller-Dieker s velkým rozsahem klinických znaků (OMIM 613215).
Více informací o těchto syndromech naleznete na adrese ncbi.nlm.nih.gov v OMIM607432 (Lissencephaly-1) a OMIM 247200 (Miller-Dieker syndrom). Fenotyp zahrnuje kortikální malformace,typické rysy obličeje a závažné neurologické abnormality.
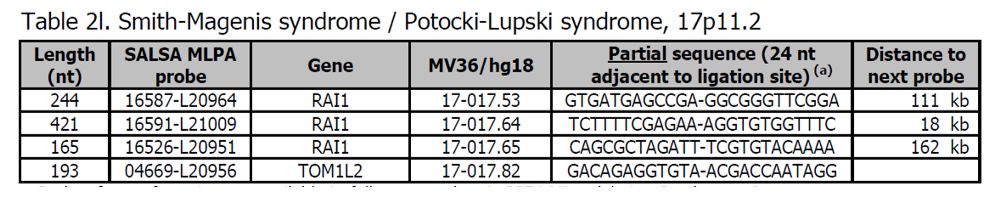 Sondy pro potvrzení jsou k dispozici v kitu P374 Microdeletion Syndromes 8. Další sondy pro oblast Smith-Magenis jsou přítomny v kitu P369 Smith-Magenis. Většina (90 %) Smith-Magenis syndromů (SMS) je způsobena intersticiální delecí 3,7 Mb na chromozomu 17p11.2. Mírnější fenotyp je spojen s duplikací stejné oblasti, což vede k Potocki-Lupskiho syndromu (PTLS).
Sondy pro potvrzení jsou k dispozici v kitu P374 Microdeletion Syndromes 8. Další sondy pro oblast Smith-Magenis jsou přítomny v kitu P369 Smith-Magenis. Většina (90 %) Smith-Magenis syndromů (SMS) je způsobena intersticiální delecí 3,7 Mb na chromozomu 17p11.2. Mírnější fenotyp je spojen s duplikací stejné oblasti, což vede k Potocki-Lupskiho syndromu (PTLS).
Více informací o SMS a PTLS naleznete na adrese ncbi.nlm.nih.gov v OMIM 182290 a v OMIM 610883. SMS je charakterizovaný výraznými fyzickými znaky, vývojovým zpožděním, kognitivním postižením, abnormalitami v chování a mírným až středně těžkým mentálním postižením. PTLS se vyznačuje hypotonií, selháním vývoje, intelektuálním postižením, pervasivními vývojovými chorobami a vrozenými anomáliemi.
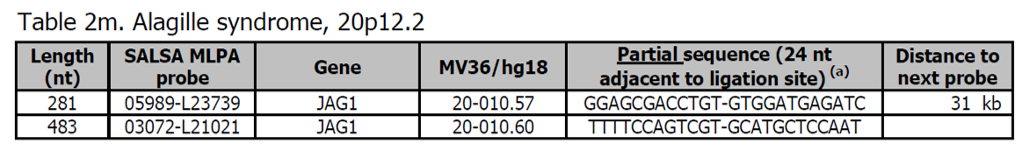
- Sondy pro potvrzení jsou k dispozici v kitu P184 JAG1.
- Většina (~ 89 %) Alagille syndromů (ALGS) je způsobena bodovou mutací genu JAG1. Přibližně 7% postižených jedinců má deleci chromozomu 20p12.2 zahrnujícího tento gen.
Více informací o Alagille syndromu najdete zde a v OMIM118450. Klinické znaky jsou vysoce variabilní a mohou zahrnovat cholestázu, vrozené srdeční vady, kosterní abnormality, oční abnormality a typické rysy obličeje.
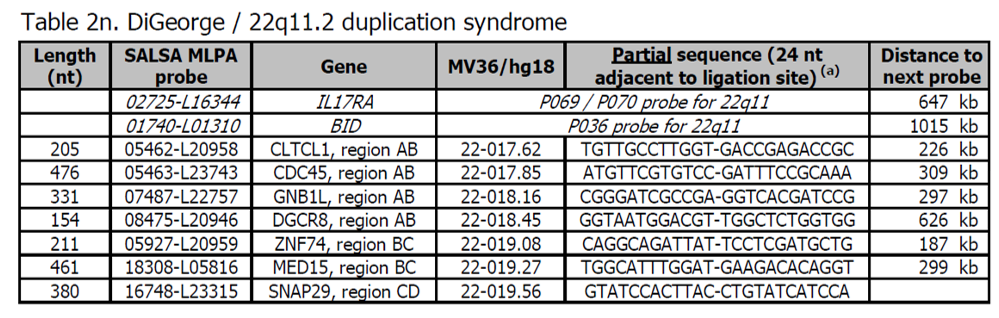
- Sondy pro potvrzení jsou k dispozici v kitu P372 Microdeletion Syndromes 6.
- Další sondy pro oblast 22q11 DiGeorge jsou přítomny v kitu P250 DiGeorge.
- Delece v 22q11 jsou nejčastější příčinou DiGeorgeova syndromu. Tyto delece 22q11 mohou mít různou velikost.Většina (~ 85%) zahrnuje oblasti AB, BC a CD, i když některé delece jsou menší (pouze AB) nebo větší.
- Pacienti se syndromem kočičího oka mohou být detekováni pomocí sond v v telomerických kitech P036 a P070, ale nikoliv sondami v tomto kitu P064.
Více informací o syndromu DiGeorge naleznete zde a v OMIM188400. Je známo, že široká škála klinických příznaků je spojena s DiGeorgeovým syndromem, včetně vrozených srdečných chorob, palatinálních abnormalit, charakteristických rysů obličeje, poruch učení, imunitní nedostatečnosti a hypokalcémie.
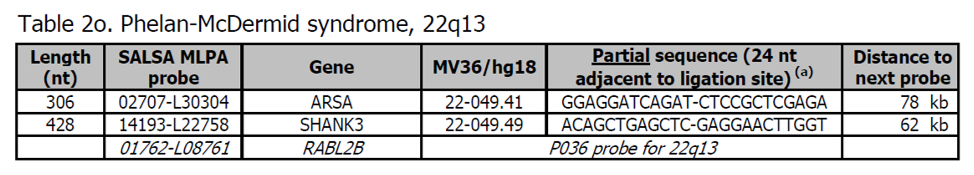
- Sondy pro potvrzení jsou k dispozici v kitu P373 Microdeletion Syndromes 7.
- Více sond v oblasti Phelan-McDermid je přítomno v kitu P188 22q13.
- Geny SHANK3 jsou podezřelé, že jsou odpovědné za alespoň část fenotypu Phelan-McDermid syndromu. Sonda RABL2B v P036 je umístěna mezi SHANK3 a 22q telomerou. Sondy ARSA v P064 a P069 / P070 detekují stejnou sekvenci.
Více informací o syndromu Phelan-McDermid naleznete zde a v OMIM 606232. Fenotyp zahrnuje neonatální hypotonii, globální vývojové zpoždění, normální až zrychlený růst, žádnou nebo výrazně zpožděnou řeč, autistické chování a drobné dysmorfické rysy. Většina jedinců má mírné až vážné intelektuální postižení.
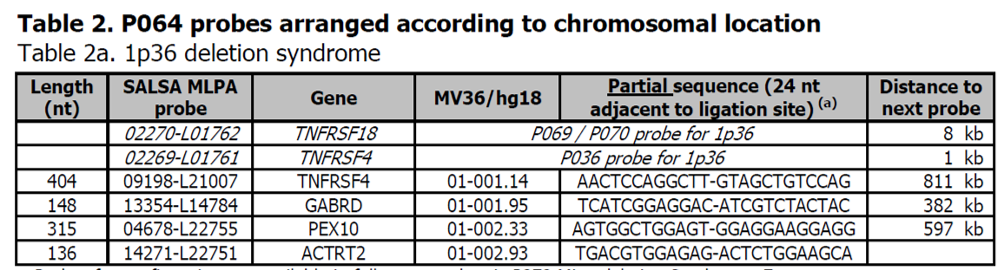
MV36 = 01-001.14 indikuje, že sonda je na chromozómu 1, ve vzdálenosti 1,14 Mb od p-teloméry podle referenční sekvence NCBI Build 36 / hg18.
Související produkty
|
|
Několik MLPA kitů pro specifické syndromy jsou k dispozici jako P250 DiGeorge, ME028 Prader-Willi/Angelman, P061 Lissencephaly a P029 WBS.
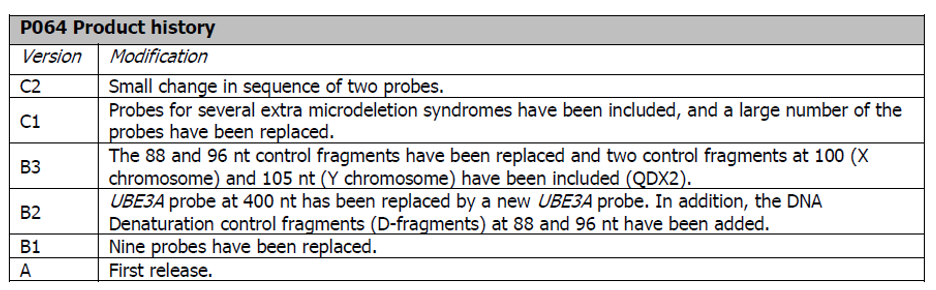
Historie produktu P064